

Why Do Mutations In Genes That Encode Dna Repair Enzymes Often Predispose An Individual To Cancer?
DNA integrity is ever nether attack from environmental agents like skin cancer-causing UV rays. How practice DNA repair mechanisms discover and repair damaged DNA, and what happens when they neglect?

Because DNA is the repository of genetic information in each living cell, its integrity and stability are essential to life. DNA, nevertheless, is not inert; rather, information technology is a chemical entity subject area to assault from the environment, and any resulting damage, if not repaired, volition lead to mutation and peradventure disease. Peradventure the all-time-known instance of the link between environmental-induced Deoxyribonucleic acid harm and illness is that of pare cancer, which can be caused past excessive exposure to UV radiations in the course of sunlight (and, to a lesser degree, tanning beds). Another case is the damage caused by tobacco smoke, which can lead to mutations in lung cells and subsequent cancer of the lung. Beyond environmental agents, DNA is as well subject to oxidative damage from byproducts of metabolism, such as free radicals. In fact, it has been estimated that an individual cell can suffer up to i million Deoxyribonucleic acid changes per twenty-four hour period (Lodish et al., 2005).
In add-on to genetic insults caused by the environs, the very procedure of DNA replication during prison cell division is prone to error. The rate at which DNA polymerase adds incorrect nucleotides during DNA replication is a major factor in determining the spontaneous mutation rate in an organism. While a "proofreading" enzyme normally recognizes and corrects many of these errors, some mutations survive this process. Estimates of the frequency at which human DNA undergoes lasting, uncorrected errors range from 1 ten 10-four to 1 x x-half dozen mutations per gamete for a given gene. A rate of 1 x 10-half dozen means that a scientist would expect to observe i mutation at a specific locus per ane million gametes. Mutation rates in other organisms are often much lower (Table 1).
One style scientists are able to estimate mutation rates is by because the charge per unit of new dominant mutations plant at different loci. For example, by examining the number of individuals in a given population who were diagnosed with neurofibromatosis (NF1, a affliction caused by a spontaneous—or noninherited—dominant mutation), scientists determined that the spontaneous mutation rate of the gene responsible for this illness averaged i 10 10-4 mutations per gamete (Crowe et al., 1956). Other researchers have found that the mutation rates of other genes, similar that for Huntington'south disease, are significantly lower than the rate for NF1. The fact that investigators have reported different mutation rates for different genes suggests that certain loci are more than prone to damage or error than others.
DNA Repair Mechanisms and Human Illness

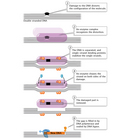
Deoxyribonucleic acid repair processes exist in both prokaryotic and eukaryotic organisms, and many of the proteins involved have been highly conserved throughout evolution. In fact, cells have evolved a number of mechanisms to detect and repair the various types of harm that tin occur to DNA, no affair whether this impairment is caused by the environment or past errors in replication. Considering DNA is a molecule that plays an active and critical office in prison cell segmentation, command of Dna repair is closely tied to regulation of the cell cycle. (Recall that cells transit through a cycle involving the G1, S, G2, and Thou phases, with DNA replication occurring in the S phase and mitosis in the M phase.) During the cell wheel, checkpoint mechanisms ensure that a cell'southward Dna is intact before permitting Dna replication and cell segmentation to occur. Failures in these checkpoints can lead to an accumulation of damage, which in turn leads to mutations.
Defects in Dna repair underlie a number of human genetic diseases that affect a wide variety of body systems just share a constellation of common traits, most notably a predisposition to cancer (Tabular array 2). These disorders include clutter-telangiectasia (AT), a degenerative motor condition caused by failure to repair oxidative damage in the cerebellum, and xeroderma pigmentosum (XP), a status characterized by sensitivity to sunlight and linked to a defect in an important ultraviolet (UV) damage repair pathway. In improver, a number of genes that accept been implicated in cancer, such as the RAD grouping, have also been determined to encode proteins critical for Deoxyribonucleic acid harm repair.
UV Damage, Nucleotide Excision Repair, and Photoreactivation

Every bit previously mentioned, one of import Dna damage response (DDR) is triggered by exposure to UV light. Of the three categories of solar UV radiation, only UV-A and UV-B are able to penetrate Earth's temper. Thus, these two types of UV radiations are of greatest business to humans, especially as standing depletion of the ozone layer causes higher levels of this radiation to achieve the planet'due south surface.
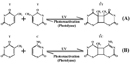
UV radiations causes two classes of DNA lesions: cyclobutane pyrimidine dimers (CPDs, Figure 1) and 6-4 photoproducts (6-4 PPs, Effigy 2). Both of these lesions distort Dna's structure, introducing bends or kinks and thereby impeding transcription and replication. Relatively flexible areas of the DNA double helix are most susceptible to impairment. In fact, one "hot spot" for UV-induced damage is constitute within a unremarkably mutated oncogene, the p53 gene.
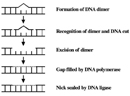
CPDs and six-4 PPs are both repaired through a procedure known as nucleotide excision repair (NER). In eukaryotes, this complex process relies on the products of approximately 30 genes. Defects in some of these genes have been shown to cause the human illness XP, every bit well equally other weather condition that share a hazard of peel cancer that is elevated about a thousandfold over normal. More specifically, eukaryotic NER is carried out past at to the lowest degree 18 poly peptide complexes via iv discrete steps (Figure 3): detection of damage; excision of the section of DNA that includes and surrounds the error; filling in of the resulting gap by Dna polymerase; and sealing of the nick betwixt the newly synthesized and older DNA (Figure 4). In bacteria (which are prokaryotes), nonetheless, the procedure of NER is completed by only three proteins, named UvrA, UvrB, and UvrC.
Leaner and several other organisms also possess another mechanism to repair UV damage chosen photoreactivation. This method is oftentimes referred to every bit "low-cal repair," because it is dependent on the presence of low-cal energy. (In comparing, NER and most other repair mechanisms are oft referred to as "dark repair," every bit they do not require lite as an energy source.) During photoreactivation, an enzyme chosen photolyase binds pyrimidine dimer lesions; in addition, a second molecule known as chromophore converts light energy into the chemical energy required to directly revert the affected expanse of DNA to its undamaged form. Photolyases are plant in numerous organisms, including fungi, plants, invertebrates such as fruit flies, and vertebrates including frogs. They do non announced to exist in humans, even so (Sinha & Hader, 2002).
Additional Deoxyribonucleic acid Repair mechanisms
NER and photoreactivation are not the only methods of DNA repair. For instance, base of operations excision repair (BER) is the predominant mechanism that handles the spontaneous Dna harm acquired by gratis radicals and other reactive species generated by metabolism. Bases can become oxidized, alkylated, or hydrolyzed through interactions with these agents. For case, methyl (CH3) chemical groups are frequently added to guanine to form vii-methylguanine; alternatively, purine groups may be lost. All such changes result in aberrant bases that must be removed and replaced. Thus, enzymes known as DNA glycosylases remove damaged bases by literally cutting them out of the Dna strand through cleavage of the covalent bonds between the bases and the sugar-phosphate backbone. The resulting gap is so filled by a specialized repair polymerase and sealed past ligase. Many such enzymes are found in cells, and each is specific to certain types of base alterations.
Yet another form of Dna damage is double-strand breaks, which are caused past ionizing radiation, including gamma rays and Ten-rays. These breaks are highly deleterious. In addition to interfering with transcription or replication, they can lead to chromosomal rearrangements, in which pieces of one chromosome become fastened to another chromosome. Genes are disrupted in this process, leading to hybrid proteins or inappropriate activation of genes. A number of cancers are associated with such rearrangements. Double-strand breaks are repaired through one of two mechanisms: nonhomologous end joining (NHEJ) or homologous recombination repair (HRR). In NHEJ, an enzyme called Dna ligase IV uses overhanging pieces of Dna adjacent to the intermission to join and fill in the ends. Additional errors tin be introduced during this procedure, which is the case if a cell has not completely replicated its DNA in preparation for division. In contrast, during HRR, the homologous chromosome itself is used as a template for repair.
Mutations in an organism's Dna are a function of life. Our genetic lawmaking is exposed to a diverseness of insults that threaten its integrity. But, a rigorous system of checks and balances is in place through the DNA repair machinery. The errors that slip through the cracks may sometimes be associated with disease, but they are also a source of variation that is acted upon by longer-term processes, such as evolution and natural pick.
References and Recommended Reading
Branze, D., & Foiani, M. Regulation of Deoxyribonucleic acid repair throughout the cell cycle. Nature Reviews Molecular Cell Biology nine, 297–308 (2008) doi:10.1038/nrm2351.pdf (link to article)
Crowe, F. W., et al. A Clinical, Pathological, and Genetic Study of Multiple Neurofibromatosis (Springfield, Illinois, Charles C. Thomas, 1956)
Lodish, H., et al. Molecular Biology of the Cell, 5th ed. (New York, Freeman, 2004)
Sinha, R. P., & Häder, D. P. UV-induced Deoxyribonucleic acid damage and repair: A review. Photochemical and Photobiological Sciences i, 225–236 (2002)

Source: https://www.nature.com/scitable/topicpage/dna-damage-repair-mechanisms-for-maintaining-dna-344/
Posted by: mullinspriont.blogspot.com
0 Response to "Why Do Mutations In Genes That Encode Dna Repair Enzymes Often Predispose An Individual To Cancer?"
Post a Comment